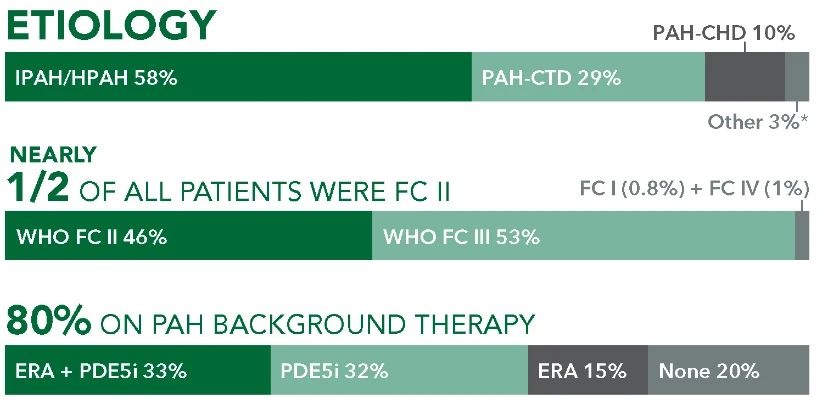
Baseline patient characteristics4
Background therapy: ERA + PDE5i
33% (n=376/1156; 31% FC II and 68% FC III)*
Etiology
- 64%
IPAH/HPAH
(n=242/376) - 26%
PAH-CTD
(n=96/376) - 5%
PAH-CHD
(n=20/376) - 5%
Other†
(n=18/376)
Time from diagnosis (mean)
- 4 years
UPTRAVI® - 3.6 years
Placebo
Time to first disease progression event in patients receiving ERA + PDE5i at baseline (UPTRAVI® vs placebo)4
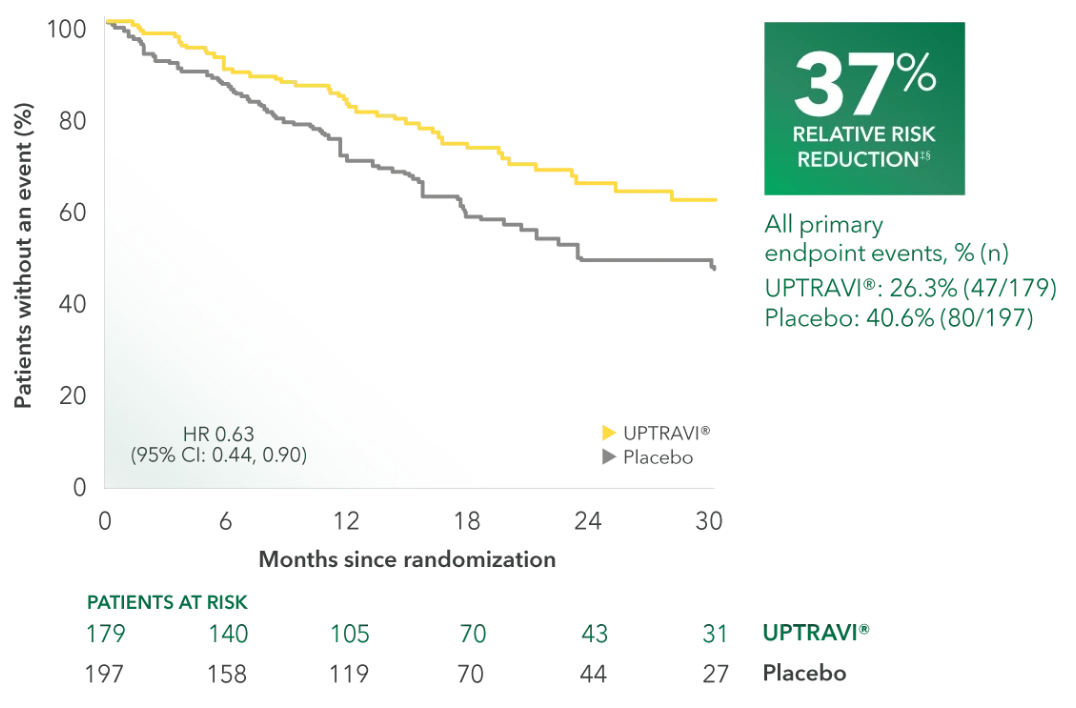
Summary of primary endpoint events in patients receiving ERA + PDE-5i at baseline4
| UPTRAVI® n=179 % (n) | Placebo n=197 % (n) | |||
|---|---|---|---|---|
| PRIMARY COMPOSITE ENDPOINT: Morbidity/mortality up to end of treatment period|| | ||||
| All primary endpoint events | 26.3% (47) | 40.6% (80) | ||
| Hospitalization for PAH | 15.1% (27) | 21.8% (43) | ||
| Other disease progression (decrease in 6MWD plus worsening FC or need for other therapy) | 6.6% (38) | 17.2% (100) | ||
| Death | 4.9% (28) | 3.1% (18) | ||
| Parenteral prostanoid or chronic oxygen therapy | 1.7% (10) | 2.2% (13) | ||
| Need for lung transplantation or balloon atrial septostomy for worsening of PAH | 0.2% (1) | 0.3% (2) | ||
Patients receiving an ERA and a PDE5i at baseline were a prespecified subgroup for evaluation of the GRIPHON primary endpoint; however, the more detailed analyses described on this page are exploratory and post hoc. Sample size should be considered and results should be interpreted with caution.
Adverse reactions in triple-combination subgroup occurring more frequently with UPTRAVI® compared with placebo by ≥3%4
| ADVERSE REACTION | UPTRAVI® n=179 % (n) | Placebo n=197 % (n) |
|---|---|---|
| Headache | 76% (136) | 39% (76) |
| Diarrhea | 56% (100) | 26% (52) |
| Nausea | 48% (85) | 25% (49) |
| Pain in jaw | 41% (74) | 10% (20) |
| Vomiting | 24% (42) | 11% (21) |
| Pain in extremity | 23% (41) | 10% (20) |
| Flushing | 20% (36) | 8% (16) |
| Myalgia | 13% (24) | 4% (8) |
| Bronchitis | 12% (21) | 9% (17) |
Baseline patient characteristics4
Background therapy in FC II patients: ERA + PDE5i
10% (n=115/1156)
Etiology
- 70%
IPAH/HPAH
(n=80/115) - 18%
PAH-CTD
(n=21/115) - 6%
PAH-CHD
(n=7/115) - 6%
Other†
(n=7/115)
Time from diagnosis (mean)
- 4.3 years
UPTRAVI® - 3.6 years
Placebo
Time to first disease progression event in FC II patients receiving ERA + PDE5i at baseline (UPTRAVI® vs placebo)4
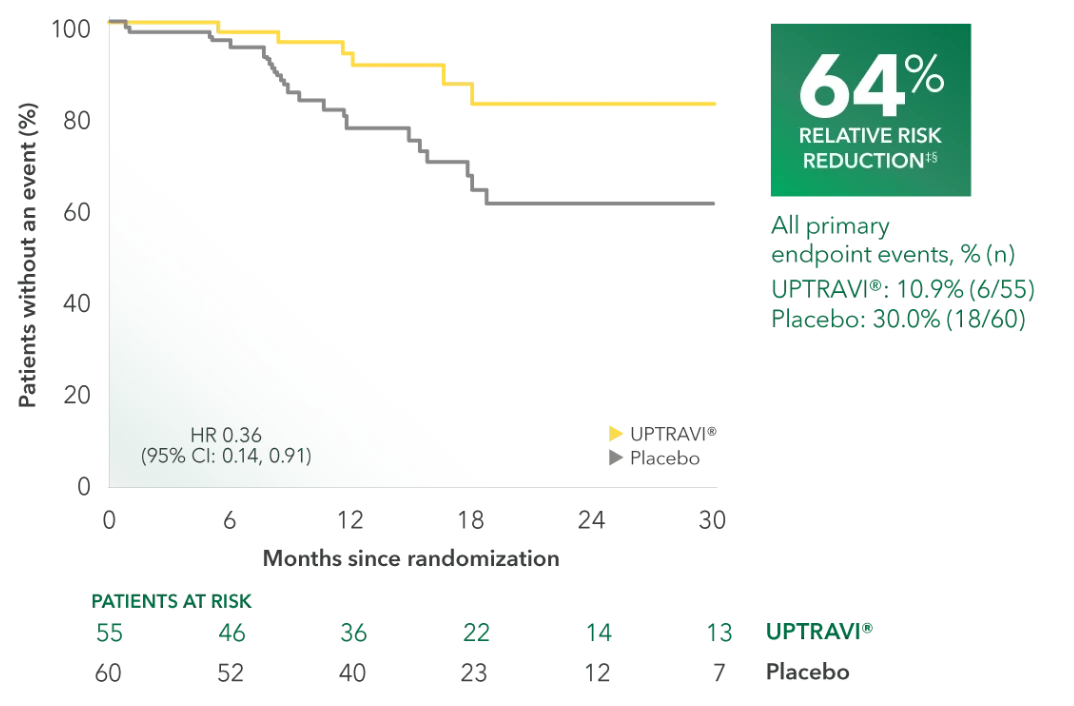
Summary of primary endpoint events in FC II patients receiving ERA + PDE5i at baseline4
| UPTRAVI® n=55 % (n) | Placebo n=60 % (n) | |||
|---|---|---|---|---|
| PRIMARY COMPOSITE ENDPOINT: Morbidity/mortality up to end of treatment period|| | ||||
| All primary endpoint events | 10.9% (6) | 30.0% (18) | ||
| Hospitalization for PAH | 5.5% (3) | 13.3% (8) | ||
| Other disease progression (decrease in 6MWD plus worsening FC or need for other therapy) | 1.8% (1) | 8.3% (5) | ||
| Death | 0% (0) | 1.7% (1) | ||
| Parenteral prostanoid or chronic oxygen therapy | 3.6% (2) | 5.0% (3) | ||
| Need for lung transplantation or balloon atrial septostomy for worsening of PAH | 0% (0) | 1.7% (1) | ||
Patients receiving an ERA and a PDE5i at baseline were a prespecified subgroup for evaluation of the GRIPHON primary endpoint; however, the more detailed analyses described on this page are exploratory and post hoc. Sample size should be considered and results should be interpreted with caution.
Adverse reactions in FC II triple-combination subgroup occurring more frequently with UPTRAVI® compared with placebo by ≥3%4
| ADVERSE REACTION | UPTRAVI® n=55 % (n) | Placebo n=60 % (n) |
|---|---|---|
| Headache | 78% (43) | 30% (18) |
| Diarrhea | 58% (32) | 13% (8) |
| Nausea | 55% (30) | 20% (12) |
| Pain in jaw | 38% (21) | 5% (3) |
| Vomiting | 24% (13) | 12% (7) |
| Pain in extremity | 15% (8) | 7% (4) |
| Flushing | 16% (9) | 8% (5) |
| Myalgia | 15% (8) | 2% (1) |
| Fatigue | 15% (8) | 10% (6) |
| Bronchitis | 13% (7) | 8% (5) |
| Abdominal pain | 15% (8) | 8% (5) |
| Decreased appetite | 11% (6) | 5% (3) |
Baseline patient characteristics4
Background therapy in FC III patients: ERA + PDE5i
22% (n=255/1156)
Etiology
- 61%
IPAH/HPAH
(n=156/255) - 29%
PAH-CTD
(n=75/255) - 5%
PAH-CHD
(n=13/255) - 4%
Other†
(n=11/255)
Time from diagnosis (mean)
- 3.9 years
UPTRAVI® - 3.6 years
Placebo
Time to first disease progression event in FC III patients receiving ERA + PDE5i at baseline (UPTRAVI® vs placebo)4
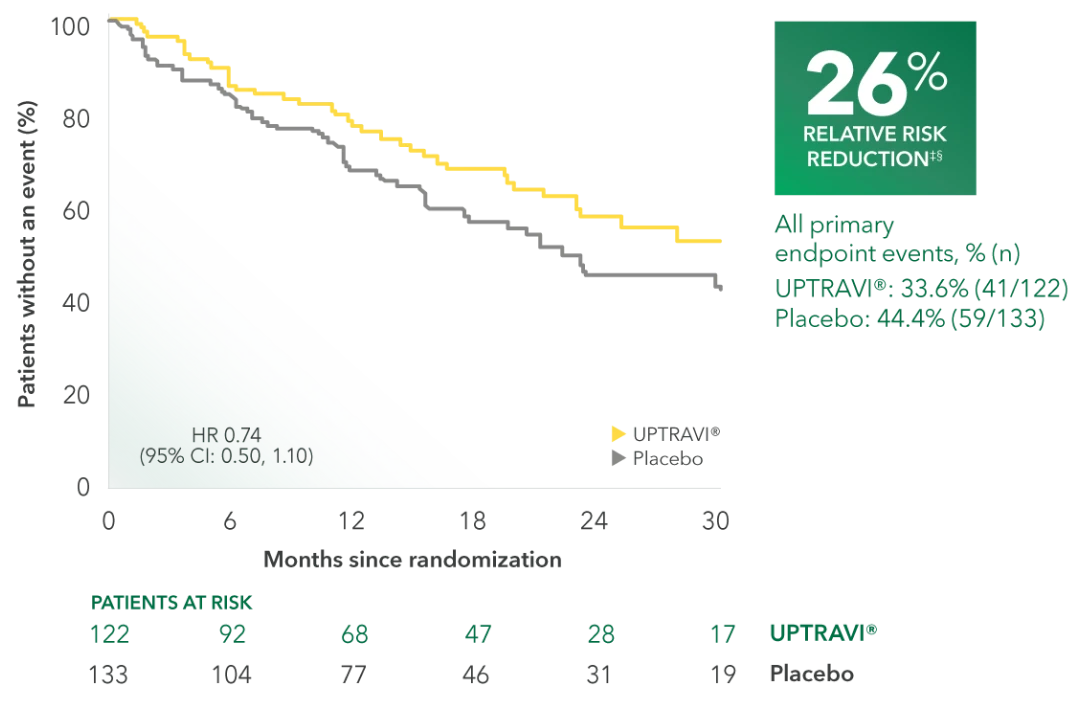
Summary of primary endpoint events in FC III patients receiving ERA + PDE5i at baseline4
| UPTRAVI® n=122 % (n) | Placebo n=133 % (n) | |||
|---|---|---|---|---|
| PRIMARY COMPOSITE ENDPOINT: Morbidity/mortality up to end of treatment period|| | ||||
| All primary endpoint events | 33.6% (41) | 44.4% (59) | ||
| Hospitalization for PAH | 19.7% (24) | 24.8% (33) | ||
| Other disease progression (decrease in 6MWD plus worsening FC or need for other therapy) | 8.2% (10) | 12.0% (16) | ||
| Death | 3.3% (4) | 1.5% (2) | ||
| Parenteral prostanoid or chronic oxygen therapy | 2.5% (3) | 5.3% (7) | ||
| Need for lung transplantation or balloon atrial septostomy for worsening of PAH | 0% (0) | 0.75% (1) | ||
Patients receiving an ERA and a PDE5i at baseline were a prespecified subgroup for evaluation of the GRIPHON primary endpoint; however, the more detailed analyses described on this page are exploratory and post hoc. Sample size should be considered and results should be interpreted with caution.
Adverse reactions in FC III triple-combination subgroup occurring more frequently with UPTRAVI® compared with placebo by ≥3%4
| ADVERSE REACTION | UPTRAVI® n=122 % (n) | Placebo n=133 % (n) |
|---|---|---|
| Headache | 75% (91) | 43% (57) |
| Diarrhea | 54% (66) | 31% (41) |
| Nausea | 43% (53) | 27% (36) |
| Pain in jaw | 43% (52) | 12% (16) |
| Vomiting | 23% (28) | 10% (13) |
| Pain in extremity | 26% (32) | 12% (16) |
| Flushing | 21% (26) | 8% (11) |
| Myalgia | 13% (16) | 5% (7) |